
The onset of ALS is often very subtle, so that the symptoms may initially be overlooked. The earliest symptoms are muscle weakness and/or muscle atrophy.
ALS does not usually present itself in a rough manner; the first symptoms, often similar to those of other diseases, can easily go unnoticed (ex. Tripping, dropping objects, trouble speaking, cramps, muscle weakness and shaking). Since there is no way of screening exclusively for ALS, it is first necessary to conduct tests to eliminate other possible illnesses. As the disease progresses, symptoms turn to muscle weakness and atrophy, meaning a decrease in muscle weight, volume and ability to function.
Over the course of ALS, certain symptoms can indicate motor neuron atrophy in the superior/ inferior portions of the dorsal spine. ALS specialists often find the following symptoms linked to motor neuron degeneration:
Inferior motor neuron degeneration:
- Muscle weakness and atrophy
- Involuntary muscle shaking
- Muscle cramps
- Weakening of reflexes
- Flaccidity (decrease in muscle tone)
- Difficulty swallowing
- Inability to properly pronounce
- Shortness of breath, even while at rest
Superior motor neuron degeneration:
- Rigidity, muscular stiffness
- Emotional lability (loss of capacity to control laughter and lacrimation
- Hyperactive reflexes
When the patient is elderly, symptoms are sometimes connected to old age. Only when muscle weakness has spread throughout the whole body can ALS finally be confirmed. The patient is then subjected to a physical exam, sometimes to an electromyogram (EMG), blood tests, magnetic resonance imaging (MRI), or other tests to find evidence of a disease resembling ALS.
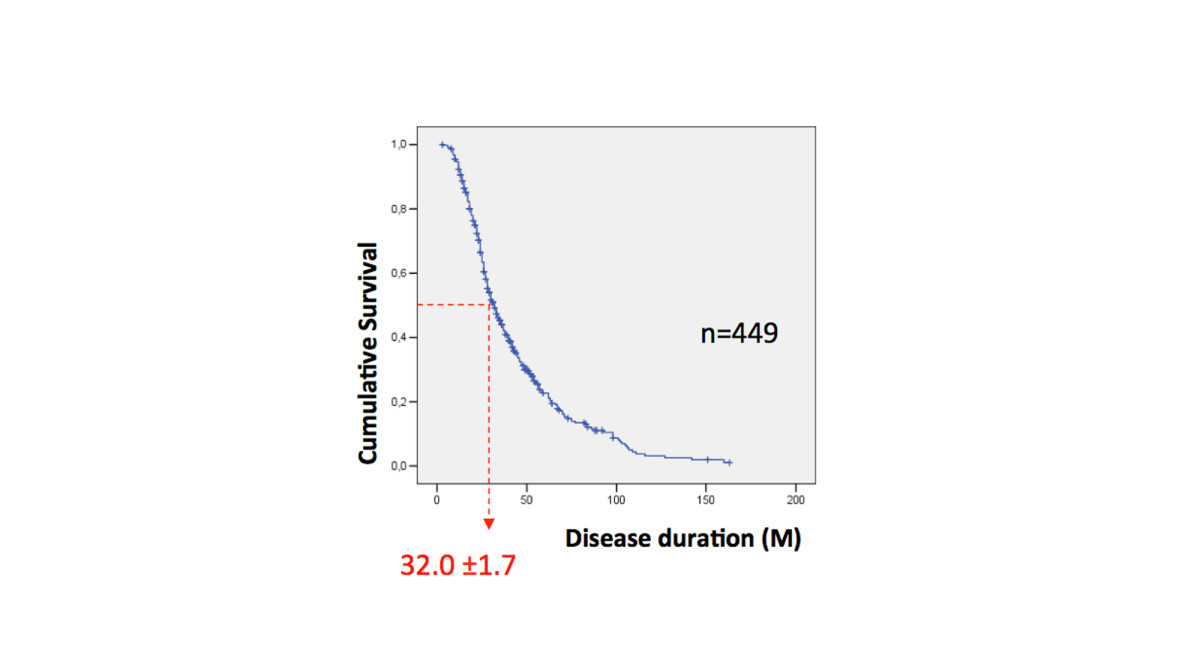
Most people with ALS die from respiratory failure, usually within 3 to 5 years from the initial onset of symptoms, although approximately 10% of patients may have a more benign form with a survival of 10 years or more.